

AI-ACCELERATED DRUG DISCOVERY
Available from Reaxense
This protein is integrated into the Receptor.AI ecosystem as a prospective target with high therapeutic potential. We performed a comprehensive characterization of Alpha-galactosidase A including:
1. LLM-powered literature research
Our custom-tailored LLM extracted and formalized all relevant information about the protein from a large set of structured and unstructured data sources and stored it in the form of a Knowledge Graph. This comprehensive analysis allowed us to gain insight into Alpha-galactosidase A therapeutic significance, existing small molecule ligands, relevant off-targets, and protein-protein interactions.
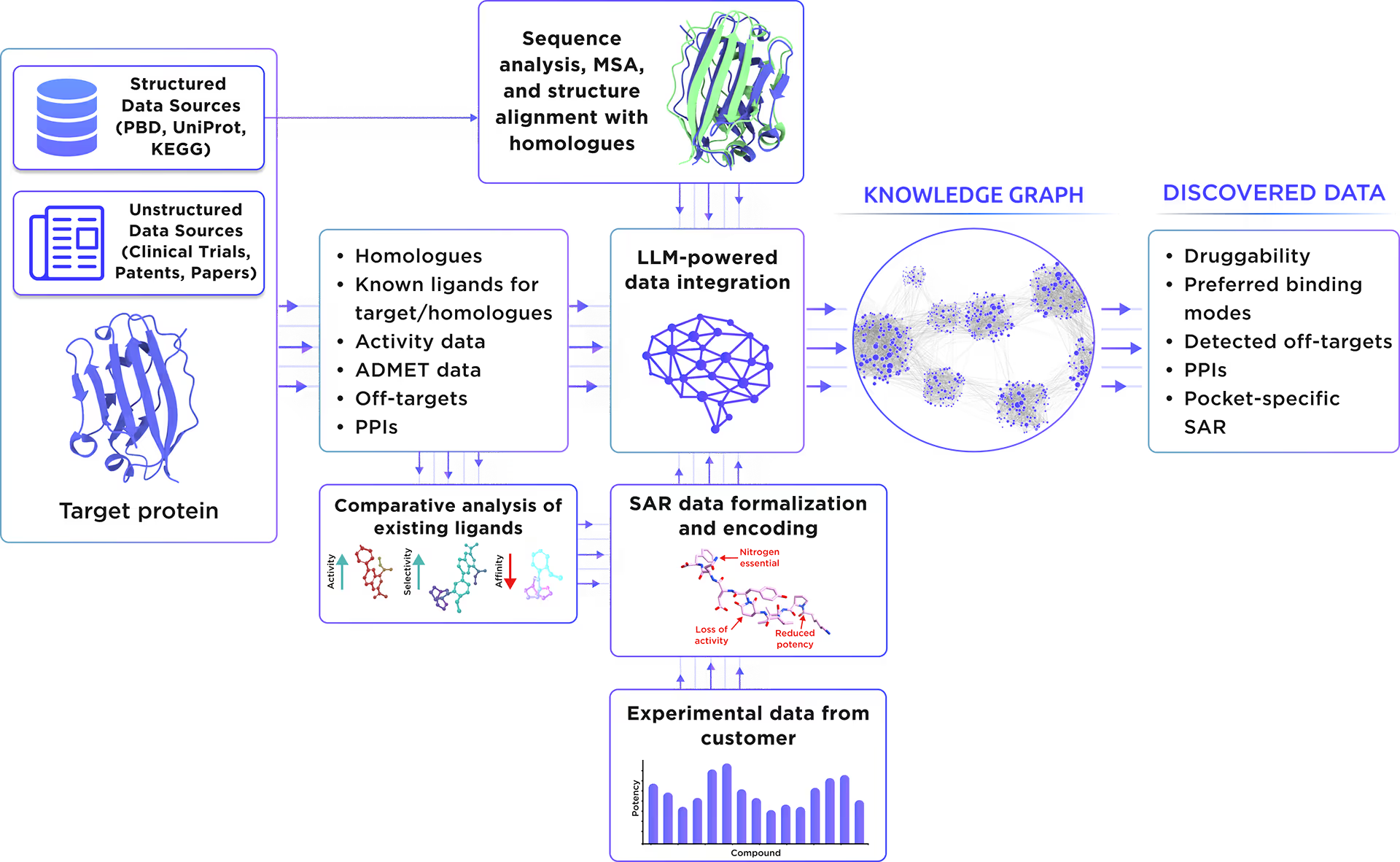
Fig. 1. Preliminary target research workflow
2. AI-Driven Conformational Ensemble Generation
Starting from the initial protein structure, we employed advanced AI algorithms to predict alternative functional states of Alpha-galactosidase A, including large-scale conformational changes along "soft" collective coordinates. Through molecular simulations with AI-enhanced sampling and trajectory clustering, we explored the broad conformational space of the protein and identified its representative structures. Utilizing diffusion-based AI models and active learning AutoML, we generated a statistically robust ensemble of equilibrium protein conformations that capture the receptor's full dynamic behavior, providing a robust foundation for accurate structure-based drug design.
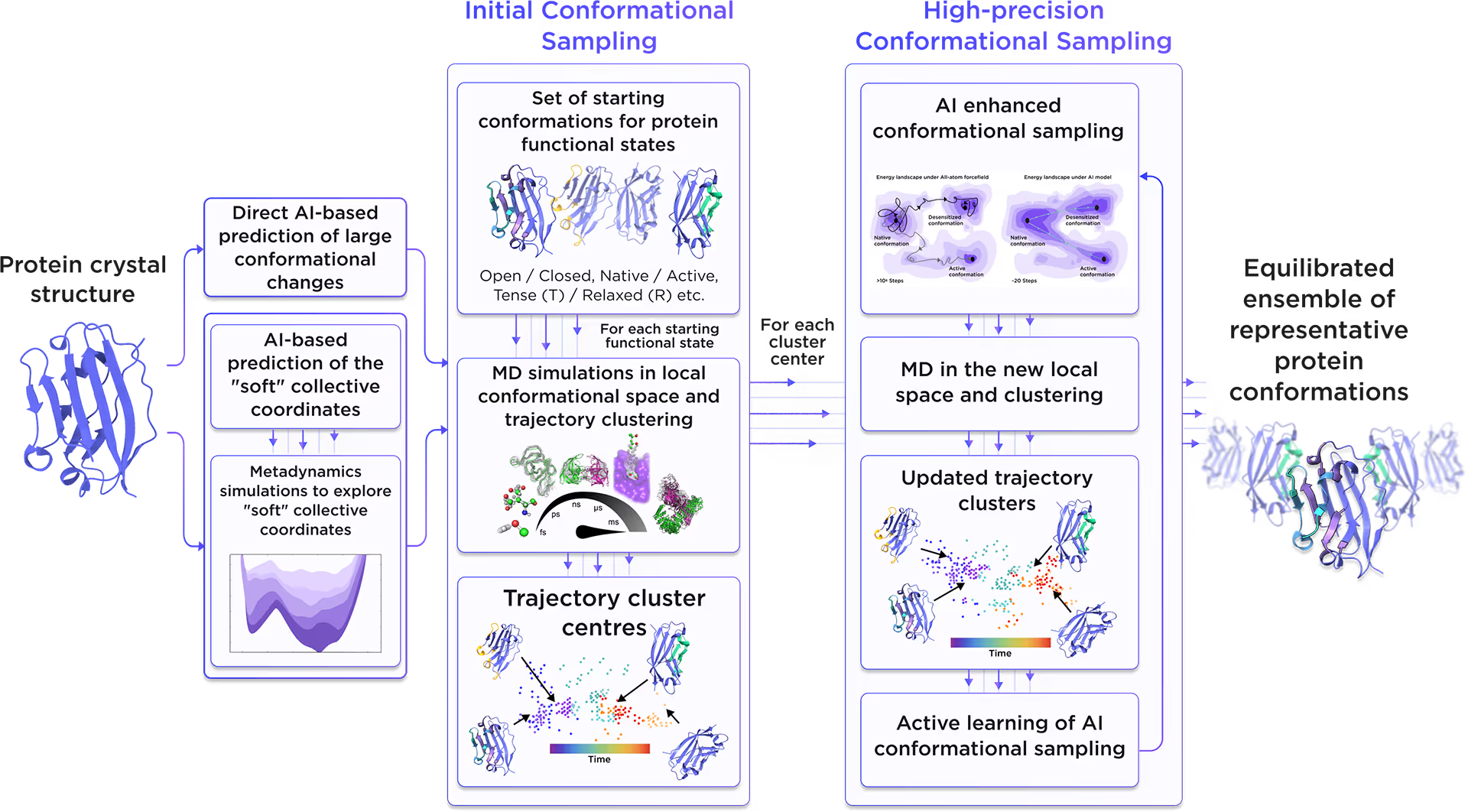
Fig. 2. AI-powered molecular dynamics simulations workflow
3. Binding pockets identification and characterization
We employed the AI-based pocket prediction module to discover orthosteric, allosteric, hidden, and cryptic binding pockets on the protein’s surface. Our technique integrates the LLM-driven literature search and structure-aware ensemble-based pocket detection algorithm that utilizes previously established protein dynamics. Tentative pockets are then subject to AI scoring and ranking with simultaneous detection of false positives. In the final step, the AI model assesses the druggability of each pocket enabling a comprehensive selection of the most promising pockets for further targeting.
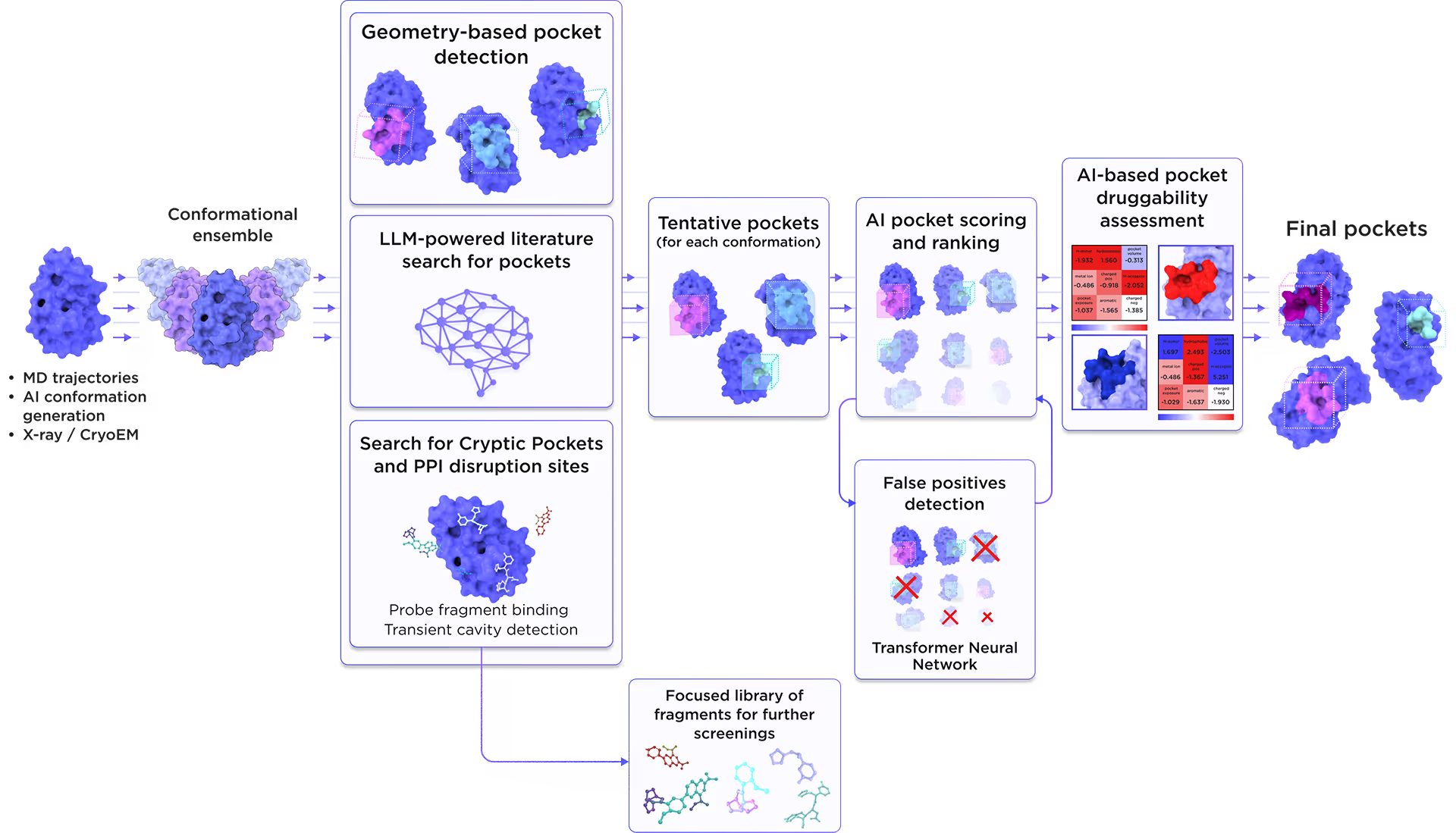
Fig. 3. AI-based binding pocket detection workflow
4. AI-Powered Virtual Screening
Our ecosystem is equipped to perform AI-driven virtual screening on Alpha-galactosidase A. With access to a vast chemical space and cutting-edge AI docking algorithms, we can rapidly and reliably predict the most promising, novel, diverse, potent, and safe small molecule ligands of Alpha-galactosidase A. This approach allows us to achieve an excellent hit rate and to identify compounds ready for advanced lead discovery and optimization.
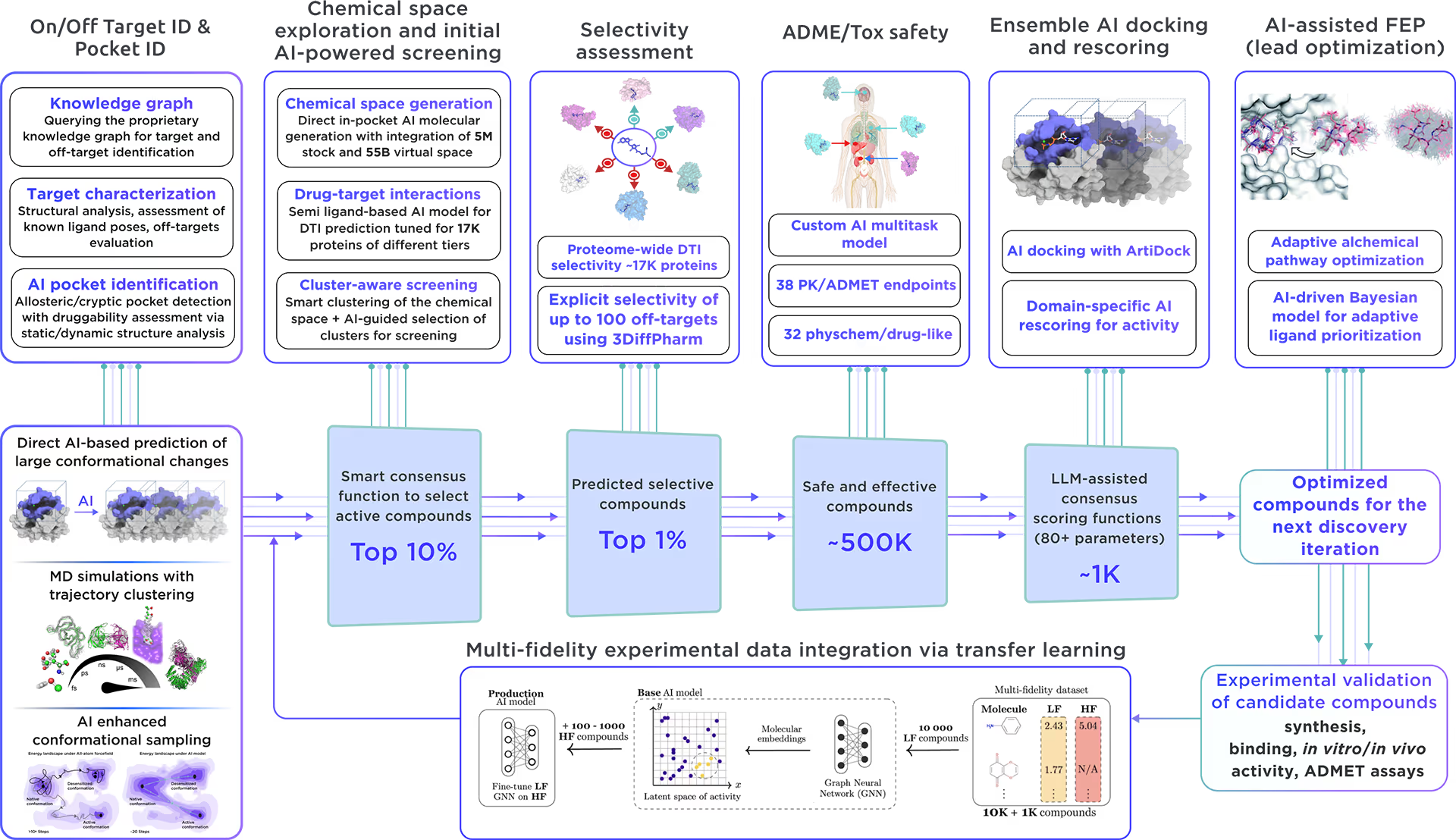
Fig. 4. The screening workflow of Receptor.AI
Receptor.AI, in partnership with Reaxense, developed a next-generation technology for on-demand focused library design to enable extensive target exploration.
The focused library for Alpha-galactosidase A includes a list of the most effective modulators, each annotated with 38 ADME-Tox and 32 physicochemical and drug-likeness parameters. Furthermore, each compound is shown with its optimal docking poses, affinity scores, and activity scores, offering a detailed summary.
Alpha-galactosidase A
partner:
Reaxense
upacc:
P06280
UPID:
AGAL_HUMAN
Alternative names:
Alpha-D-galactosidase A; Alpha-D-galactoside galactohydrolase; Galactosylgalactosylglucosylceramidase GLA; Melibiase
Alternative UPACC:
P06280; Q6LER7
Background:
Alpha-galactosidase A, also known as Alpha-D-galactosidase A, Alpha-D-galactoside galactohydrolase, Galactosylgalactosylglucosylceramidase GLA, and Melibiase, plays a crucial role in the lysosomal degradation of glycosphingolipids. This enzyme's activity is pivotal for the breakdown and removal of glycolipids from the body, ensuring cellular health and proper metabolic functions.
Therapeutic significance:
The deficiency of Alpha-galactosidase A leads to Fabry disease, a rare X-linked sphingolipidosis characterized by the systemic accumulation of globotriaosylceramide. This accumulation causes a range of symptoms, including skin lesions, ocular deposits, and renal failure. Understanding the enzyme's function and its genetic variants opens avenues for targeted gene therapy and enzyme replacement therapies, offering hope for patients with Fabry disease.